Estudio INA 1 . 2 . . observaciones 1 . 2 . 3 . . planicie . . ley de humedales . 1 . 2 . 3 . 4 . 5 . 6 . 7 . 8 . 9 . 10 . 11 . 12 . 13 . 14 . . Salida Luján 1 . 2 . 3 . 4 . 5 . 6 . 7 . . Parque Industrial 1 . 2 . 3 . . Larena . . Aliviador . . Vinculación . 1 . 2 . . Pilará 1 . 2 . 3 . 4 . 5 . 6 . 7 . 8 . 9 . 10 . 11 . 12 . 13 . . causa Pilará 1 . 2 . 3 . 4 . 5 . 6 . 7 . 8 . 9 . . planEscobar 1 . 2 . 3 . 4 . . Ord 727 1 . 2 . 3 . . Consultatio 1 . 2 . 3 . 4 . 5 . 6 . 7 . 8 . 9 . 10 . 11 . 12 . . altimetrias . . San Sebastián 1 . 2 . 3 . 4 . 5 . 6 . 7 . 8 . 9 . 10 . 11 . 12 . 13 . 14 . 15 . 16 . 17 . 18 . 19 . 20 . 21 . 22 . 23 . 24 . 25 . 26 . 27 . 28 . 29 . . embalses . . EIDICO . 1 . 2 . . mentiras . . quantum . . El cazal EIRSA 1 . 2 . 3 . 4 . 5 . . La Cañada 1 . 2 . . humedal Escobar 1 . 2 . 3 . . Cartas Doc a Scioli . 1 . 2 . 3 . 4 . 5 . . miserias . . cartas doc al OPDS 1 . 2 . 3 . a la AdA . al Juzg Fed 1ºSI . al Fiscal Federal . a Sergio Massa 1 . 2 . . a Zúccaro 1 . 2 . 3 . . a Arlía . . a Alvarez Rodríguez 1 . 2 . 3 . . a Ma Eug. Vidal . . a otros . . atropellos 1 . 2 . 3 . 4 . . playboy . 1 . 2 . . puertoescobar 1 . 2 . 3 . 4 . . areco . . cloaca 1 . 2 . 3 . . causa 2843 JF1SI 1 . 2 . 3 . 4 . 5 . . Colony Park 1 . 2 . . preguntas 1 . 2 . 3 . 4 . 5 . . respuestas . . remediacion . . recusacion . . amicus . . propuesta 1 . 2 . . terraplen . . jurisprud . . archivolegislativos . . hidrolinea 1 . 2 . 3 . 4 . . . art 59 . . Res.29/09 . . eiaydia 1 . 2 . 3 . 4 . 5 . 6 . 7 . 8 . 9 . 10 . . Valls . . parentescos . . contralor . . salvedades . . IAB . . flujo termodinámico 1 . 2 . 3 . convenglish . . plataforma 1 . 2 . . termodinamica 1 . 2 . 3 . . riovivo . . riomuerto . . mantos . . sedimentología . . acuíferos . . puelches 1 . 2 . . albanueva . . sustentable. 1 . 2 . 3 . 4 . 5 . 6 . 7 . 8 . 9 . 10 . 11 . . agua 1 . 2 . 3 . . pendientes 1 . 2 . . Luján . 1 . 2 . 3 . maná del cielo 1 . 2 . 3 . . ensanche . 1 . 2 . 3 . 4 . 5 . 6 . 7 . 8 . 9 . 10 . 11 . 12 . 13 . 14 . 15 . 16 . 17 . 18 . 19 . 20 . . Zanjón Villanueva . 1 . 2 . 3 . . garin . . cantón . . las tunas . . ley particular . . emergencias . 1 . 2 . inundate . 1 . 2 . 3 . 4 . 5 . . colinacarmel . . carmel . . Comilú . 1 . 2 . 3 . . comireclu . . otamendi . . Verazul . 1 . 2 . 3 . . Anibal . . jubileo . . cauce robado . 1 . 2 . 3 . 4 . 5 . 6 . . hidrometrias . . invitacion . . linea de ribera . 1 . 2 . 3 . 4 . 5 . . cartadocdevido . . compuertas . . serman . 1 . 2 . 3 . 4 . 5 . 6 . 7 . . audienciaremeros . . Silvosa . . venice . . canalsantamaria . . martindelaisla . . edafologias . . index

¿Es acaso el agua sólo materia; o sólo energía; o ambas cosas a la vez?
¿Es acaso espíritu, o acaso raíz. Es acaso cimiento; acaso savia?
¿Depende ella de nosotros; o nosotros de ella?
¿Qué toma ella de nosotros; y qué, nosotros de ella?
¿A quién pertenecen sus cauces: al dueño de la heredad; al dominio público; o a ella?
¿Acaso tiene ella entidad como para pertenecerle algo?
¿En qué cosmovisión cabe esta pregunta?
¿Qué nos enseña la füsis cuántica? ¿Es acaso viable su acceso a través de analogía?
¿Y si no lo fuera, cómo accederíamos? ¿Si con la razón no fuera, alcanzaría una caricia?
Si el bit cuántico nos habla, no de uno u otro; no de uno o cero por separado; sino de ambos a la vez; ¿cómo acceder con la razón a ello?
Si tan sólo la millonésima ava parte de la materia fuera lo que sospechamos; y el resto que la presenta fuera vacío; o algo que ni siquiera lograríamos nombrar porque la misma palabra vacío sólo regalaría sospecha de lo que no sospechamos;
qué inconveniente habría de localizar allí al cero; siendo que allí tal vez encontraría pareja que le diera Vida y fecundidad.
Si lo que brota fuera fruto de esa unión misteriosa del uno y el cero, qué inconveniente habría de cambiar un poquito nuestra cosmovisión y nuestros abordajes racionales.
Hoy está bien probado que sólo una inteligencia artificial parece en condiciones de relacionarse con los procesos de esta füsis que la razón e inteligencia humana, en parálisis completa de sus procesos delata.
Si en 25 años tendremos las primeras computadoras cuánticas operando entre nosotros, qué haremos con nuestros criterios jurisprudenciales que afirman el derecho por medio de una razón que habrá quedado perdida de su recurso analógico, frente a la enorme confianza y responsabilidad que la füsis cuántica reclama para darnos hospedaje en sus hoy crecidos misterios.
De pronto nos veremos tan pequeños, que dudo queden conceptos afines a los que hasta hoy a la cibernética asistieron.
Por ello, imaginar que pudiera merecer el apelativo de “hermenéutico”, la tarea de sopesar el peso y lugar de las palabras rescatadas de contextos con el mayor esmero, ya me resulta inmerecido premio a una labor que sólo usó de la razón para tallar verdad en un lugar en extremo estrecho;
y tan rígido, que ni siquiera a poesía ni a abismo convendría; siendo que es aquí donde la inspiración hermenéutica florecería para anticipar ese nuevo día en que la razón se rindió a cuidado amoroso.
El amor de los cauces que las aguas reflejan me hacen imposible imaginarlas que una u otro me pertenecieran.
Más bien siento que agradeceré ser amigo de los cauces y de las aguas que los riegan.
Tener dominio sobre una u otro, me parece una ilusión que no lograría sostener en intimidad sincera. Me bastaría con ser inquilino.
Permanecer junto a ellos me incorporaría sus Vidas; me descubriría sus tesoros, me alegraría los días. Y heredaría esa alegría para sembrar en mis descendencias.
Son esos los tesoros que aprecia el Hombre de campo en su soledades, que no quisiera ver interrumpidas.
Privacidad obligada de tantas Vidas, que tal vez un día empiecen a reconocer perdida. Y será de lamentar.
Pero hay acaso forma de evitar ese proceso, otro que no sea la paulatina prudencia y la gradual apertura en una dirección, que a qué dudar, luce inevitable.
Tal vez ese día, cultura sea la intermediaria, frente a tantas dudas.
Cultura es la llave que abre las puertas del hospedaje humano. Y esta füsis cuántica debe tener preparada alguna sorpresa para activar una nueva e incomparable cultura.
La palabra dominio quedará perdida en los tiempos. La palabra cuidado será la que inaugure garantía.
Cuántos pueblos hay que no conocen el dominio de la Tierra, y sin embargo la veneran.
La palabra seguridad, la palabra poder, la palabra dominio, no tienen nada que hacer en estos nuevos tiempos que la füsis cuántica inaugura.
La sinceridad interior y los comportamientos que en ella se cultivan, son la amplísima vía que no habrá de esperar a que la crean. Ya está creada.
Sólo nos resta, vivir acorde para estar a tiro de descubrirla, con la confianza natural del que no necesita esperar ese día.
No es un código de emperadores el que guía. Ni una justicia de poderosos que somete a sus sometidos al dios de sus sistemas.
Sino el paso a paso natural del que ya venera su habitat y le cuida.
Esta füsis ama el ocultarse, pues es inmanencia pura. Y si trasciende, es a través de los que la acarician en sus ánimos y en sus actos diarios.
En el agua más inmunda hoy caminan nuestras miserias. Todos los ríos y todos los arroyos y todos los arroyitos son camino de nuestras miserias.
Que no van embarcadas en barcas de una tonelada, sino en barcas invisibles,
floculando centenares de miles de toneladas de miserias que nadie las lleva,
sino esa beatífica energía del agua y lixiviada materia que la encauza, en un siglo que ha multiplicado en sus fondos y riberas, extendidas coalescensias.
Del emisario de hidrocarburos que aflora en la isla Lucha, ya han ido en camino al océano 10 Exxon Valdéz de miserias;
acumulando coalescencias en enormes áreas a punto de aflorar avulsiones centenarias.
Y todos enterados; desde conjueces de la Suprema, hasta secretarias de la Secretaria; y nadie habla.
No busquemos la barca en otro lado para seguir ocultando nuestras miserias.
Descubramos esa barca en nuestros flujos, en sus disociaciones, en sus frenos, en sus dormiciones; en toda esta miseria que descubre nuestra dominialidad y que ninguna escritura ni agrimensura revelan.
Cuando de hecho, a centímetros de la planta de los pies de poderosos señores del delta Norte, están definitivamente estancadas estas miserias.
¿De qué hablamos cuando adquirimos una de estas parcelas? ¿Sólo de lo que está a la vista? ¿Quién resistiría enterarse dónde están parados?
Riberas las hay por doquier. Y es probable que hasta que no comencemos a tirar líneas en las riberas del alma, sea inútil querer garantizar dominios.
Las dificultades en los registros dominiales tal vez sean providencias para querer lo que nos es dado, de manera más entrañable; y así poética; y así sincera.
Francisco Javier de Amorrortu . 23 de Mayo del 2007
![]()
Yendo al grano, / 26 de Agosto del 2011
All know that the drop merges into the ocean but few know that the ocean merges into the drop. -Kabir, reformer, poet (late 15th century).
"Lo que sabemos es una gota de agua; lo que ignoramos es el océano". Isaac Newton
"[Es] “una de las sustancias químicas más investigadas, pero sigue siendo la menos entendida”.
“No hay nada cuyo comportamiento sea tan complejo”.
“H20 debería ser un gas, [...] pero es un líquido. Además, cuando se congela [...] y pasa al estado sólido, el hielo flota en lugar de hundirse”.
Escritor de divulgación científica del London Imperial College
En 1916, el químico americano Gilbert Newton Lewis propusó que los enlaces químicos se formaban entre los átomos porque los electrones de los átomos interactuaban entre ellos. Lewis había observado que muchos elementos eran más estables cuando ellos contenían ocho electrones en su envoltura de valencia. El sugirió que los átomos con menos de ocho valencias de electrones se enlazaban para compartir electrones y completar sus envolturas de valencia.
Su trabajo estableció la base de lo que se conoce hoy en día sobre los enlaces químicos. Sabemos que hay dos principales tipos de enlaces químicos, iónicos y enlaces covalentes.
Enlaces Iónicos
En los enlaces iónicos, los electrones se transfieren completamente de un átomo a otro. Durante este proceso de perder o ganar electrones cargados negativamente, los átomos que reaccionan forman iones. Lo iones cargados de manera opuesta se atraen entre ellos a través de fuerzas electroestáticas que son la base del enlace iónico.
Por ejemplo, durante la reacción del sodio con el cloro:

el sodio (a la izquierda) pierde su única valencia de electrones al cloro (a la derecha), resultando en:

un ión de sodio cargado positivamente (izquierda) y un ión de cloro cargado negativamente (derecha).
Note que cuando el sodio pierde su electrón de valencia, se hace más pequeño, mientras que el cloro se hace más grande cuando gana una valencia de electrón adicional. Esto es típico de los tamaños relativos de iones a átomos. Después que la reacción tiene lugar, los iones cargado Na+ y Cl- se sujetan gracias a las fuerzas electroestáticas, formando así un enlace ionico. Los compuestos iónicos comparten muchas caractéristicas en común:
- Los enlaces iónicos se forman entre metales y no metales.
- Los compuestos iónicos se disuelven facilmente en el agua y otros solventes polares.
- En una solución, los compuestos iónicos fácilmente conducen electricidad.
- Los compuestos iónicos tienden a formar sólidos cristalinos con temperaturas muy altas.
Esta última característica es un resultado de las fuerzas intermoleculares (fuerzas entre las moléculas) en los sólidos iónicos.
El concepto de una sola molécula no aplica a cristales iónicos porque el sólido existe como un sistema continuo. Sólidos iónicos forman cristales con altos puntos de fusion debido a las a las grandes fuerzas entre dos iones vecinos.
Enlaces Covalentes
El segundo mayor tipo de enlace atómico ocurre cuando los átomos comparten electrones. Al contrario de los enlaces iónicos en los cuales ocurre una transferencia completa de electrones, el enlace covalente ocurre cuando dos (o más) elementos comparten electrones. El enlace covalente ocurre porque los átomos en el compuesto tienen una tendencia similar hacia los electrones (generalmente para ganar electrones). Esto ocurre comúnmente cuando dos no metales se enlazan. Ya que ninguno de los no elementos que participan en el enlace querrán ganar electrones, estos elementos compartirán electrones para poder llenar sus envolturas de valencia. Un buen ejemplo de un enlace covalente es ese que ocurre entre dos átomos de hidrógeno. Los átomos de hidrógeno (H) tiene un electrón de valencia en su primera envoltura. Puesto que la capacidad de esta envolutura es de dos electrones, cada átomo hidrógeno 'querrá' recoger un segundo electrón. En un esfuerzo por recoger un segundo electrón, el átomo de hidrógeno reaccionará con átomos H vecinos para formar el compuesto H2. Ya que el compuesto de hidrógeno es una combinación de átomos igualados, los átomos compartirán cada uno de sus electrones individuales, formando así un enlace covalente. De esta manera, ambos átomos comparten la estabilidad de una envoltura de valencia.
Ya que los electrones están compartidos en molécula covalentes, no se forman cargas iónicas. Por consiguiente, no hay fuerzas intermoleculares fuertes en los compuestos covalentes tal como las hay en las moléculas iónicas. Como resultado, muchos compuestos iónicos son gases o líquidos a temperatura ambiente en vez de sólidos como los compuestos iónicos en las moléculas covalentes que tienden a tener una atracción intermolecular más debil. Igualmente, al contrario de los compuestos iónicos, los compuestos covalentes existen como verdaderas moléculas.
Enlaces Múltiples: Para cada par de electrones compartidos entre dos átomos, se forma un enlace covalente único. Algunos átomos pueden compartir múltiples pares de electrones, formando enlaces covalentes múltiples. Por ejemplo, el oxígeno (que tiene seis electrones de valencia) necesita dos electrones para completar su envoltura de valencia. Cuando dos átomos de oxígeno forman el compuesto O2, ellos comparten dos pares de electrones, formando dos enlaces covalentes.
Enlaces Polares y No-Polares
En realidad, hay dos sub tipos de enlaces covalente. La molécula H2 es un buen ejemplo del primer tipo de enlace covalente el enlace no polar. Ya que ambos átomos en la molécula H2 tienen una igual atracción (o afinidad) hacia los electrones, los electrones que se enlazan son igualmente compartidos por los dos átomos, y se forma un enlace covalente no polar. Siempre que dos átomos del mismo elemento se enlazan, se forma un enlace no polar .
Un enlace polar se forma cuando los electrones son desigualmente compartidos entre dos átomos. Los enlaces polares covalentes ocurren porque un átomo tiene una mayor afinidad hacia los electrones que el otro (sin embargo, no tanta como para empujar completamente los electrones y formar un ión). En un enlace polar covalente, los electrones que se enlazan pasarán un mayor tiempo alrededor del átomo que tiene la mayor afinidad hacia los electrones. Un buen ejemplo del enlace polar covalente es el enlace hidrógeno - oxígeno en la molécula de agua.

Las moléculas de agua contienen dos átomos de hidrógeno (dibujados en rojo) enlazados a un átomo de oxígeno (en azul). El oxígeno, con seis electrones de valencia, necesita dos electrones adicionales para completar su envoltura de valencia. Cada hidrógeno contiene un electrón. Por consiguiente el oxígeno comparte los electrones de dos átomos de hidrógeno para completar su propia envoltura de valencia, y en cambio, comparte dos de sus propios electrones con cada hidrógeno, completando la envoltura de valencia H.
Enlace polar covalente simulado en una molécula de agua
La principal diferencia entre el enlace H-O en el agua y el enlace H-H, es el grado de los electrones compartidos. El gran átomo de oxígeno tiene una mayor afinidad hacia los electrones que los pequeños átomos de hidrógeno. Ya que el oxígeno tiene una atracción más fuerte en los electrones que se enlazan, el electrón ocupado anteriormente conduce a una desigual participación
Los Dipolos
Ya que los electrones de valencia en las moléculas de agua ocupan más tiempo alrededor del átomo de oxígeno que los átomos de hidrógeno, la parte de oxígeno de la molécula desarrolla una carga parcial negativa (debido a la carga negativa en los electrones). Por la misma razón, la parte de hidrógeno de la molécula desarrolla una carga parcial positiva. Los iones no se forman, a pesar de que la molécula desarrolla en su interior una carga eléctrica parcial llamada un dipolar. El dipolo de agua está representado por una flecha en la animación en la cual la cabeza de la flecha apunta hacia la parte densa final (negativa) del electrón del dipolo y el otro electrón se ecuentra cerca de la parte delgada final (positiva) al otro lado de la molécula.
Henry Cavendish descubrió en 1781 que el agua es una sustancia compuesta y no un elemento, como se pensaba desde la Antigüedad. Los resultados de dicho descubrimiento fueron desarrollados por Antoine Laurent de Lavoisier dando a conocer que el agua estaba formada por oxígeno e hidrógeno. En 1804, el químico francés Joseph Louis Gay-Lussac y el naturalista y geógrafo alemán Alexander von Humboldt publicaron un documento científico que demostraba que el agua estaba formada por dos volúmenes de hidrógeno por cada volumen de oxígeno (H2O).
Entre las moléculas de agua se establecen enlaces por puentes de hidrógeno debido a la formación de dipoloselectrostáticos que se originan al situarse un átomo de hidrógeno entre dos átomos más electronegativos, en este caso de oxígeno. El oxígeno, al ser más electronegativo que el hidrógeno, atrae más, hacia este, los electrones compartidos en los enlaces covalentes con el hidrógeno, cargándose negativamente, mientras los átomos de hidrógeno se cargan positivamente, estableciéndose así dipolos eléctricos. Los enlaces por puentes de hidrógeno son enlaces por fuerzas de van der Waals de gran magnitud, aunque son unas 20 veces más débiles que los enlaces covalentes.
Los enlaces por puentes de hidrógeno entre las moléculas del agua pura son responsables de la dilatación del agua al solidificarse, es decir, su disminución de densidad cuando se congela. En estado sólido, las moléculas de agua se ordenan formando tetraedros, situándose en el centro de cada tetraedro un átomo de oxígeno y en los vértices dos átomos de hidrógeno de la misma molécula y otros dos átomos de hidrógeno de otras moléculas que se enlazan electrostáticamente por puentes de hidrógeno con el átomo de oxígeno. La estructura cristalina resultante es muy abierta y poco compacta, menos densa que en estadolíquido. El agua tiene una densidad máxima de 1 g/cm³ cuando está a una temperatura de 4 °C,[2] característica especialmente importante en la naturaleza que hace posible el mantenimiento de la vida en medios acuáticos sometidos a condiciones exteriores de bajas temperaturas.
La dilatación del agua al solidificarse también tiene efectos de importancia en los procesos geológicos de erosión. Al introducirse agua en grietas del suelo y congelarse posteriormente, se originan tensiones que rompen las rocas.
Disolvente
El agua es descrita muchas veces como el solvente universal, porque disuelve muchos de los compuestos conocidos. Sin embargo, no lo es (aunque es tal vez lo más cercano), porque no disuelve a todos los compuestos y, de hacerlo, no sería posible construir ningún recipiente para contenerla.
El agua es un disolvente polar, más polar, por ejemplo, que el etanol. Como tal, disuelve bien sustancias iónicas y polares, como la sal de mesa (cloruro de sodio). No disuelve, de manera apreciable, sustancias fuertemente apolares, como el azufre en la mayoría de sus formas alotrópicas, además, es inmiscible con disolventes apolares, como el hexano. Esta cualidad es de gran importancia para la vida.
Esta selectividad en la disolución de distintas clases de sustancias se debe a su capacidad para formar puentes de hidrógeno con otras sustancias que pueden presentar grupos polares, o con carga iónica, como: alcoholes, azúcares con grupos R-OH, aminoácidos y proteínas con grupos que presentan cargas parciales + y − dentro de la molécula, lo que da lugar a disoluciones moleculares. También, las moléculas de agua pueden disolver sustancias salinas que se disocian formando disoluciones iónicas.
En las disoluciones iónicas, los iones de las sales orientan, debido al campo eléctrico que crean a su alrededor, a los dipolos del agua, quedando "atrapados" y recubiertos de moléculas de agua en forma de iones hidratados o solvatados.
Algunas sustancias, sin embargo, no se mezclan bien con el agua, incluyendo aceites y otras sustancias hidrofóbicas. Membranas celulares, compuestas de lípidos y proteínas, aprovechan esta propiedad para controlar las interacciones entre sus contenidos químicos y los externos, lo que se facilita, en parte, por la tensión superficial del agua.
La capacidad disolvente es responsable de:
- Las funciones metabólicas.
- Los sistemas de transporte de sustancias en los organismos.
Polaridad
La molécula de agua es muy polar, puesto que hay una gran diferencia de electronegatividad entre el hidrógeno y el oxígeno. Los átomos de oxígeno son mucho más electronegativos (atraen más a los electrones) que los de hidrógeno, lo que dota a los dos enlaces de una fuerte polaridad eléctrica, con un exceso de carga negativa del lado del oxígeno, y de carga positiva del lado de los hidrógenos.

Los dos enlaces no están opuestos, sino que forman un ángulo de 104,45° debido a la hibridación sp3 del átomo de oxígeno así que, en conjunto, los tres átomos forman un molécula angular, cargado negativamente en el vértice del ángulo, donde se ubica el oxígeno y, positivamente, en los extremos de la molécula, donde se encuentran los hidrógenos.

Este hecho tiene una importante consecuencia, y es que las moléculas de agua se atraen fuertemente, adhiriéndose por donde son opuestas las cargas. En la práctica, un átomo de hidrógeno sirve como puente entre el átomo de oxígeno al que está unido covalentemente y el oxígeno de otra molécula. La estructura anterior se denomina enlace de hidrógeno o puente de hidrógeno.

El hecho de que las moléculas de agua se adhieran electrostáticamente, a su vez modifica muchas propiedades importantes de la sustancia que llamamos agua, como la viscosidad dinámica, que es muy grande, o los puntos (temperaturas) de fusión y ebullición o los calores de fusión y vaporización, que se asemejan a los de sustancias de mayor masa molecular.
Cohesión
La cohesión es la propiedad con la que las moléculas de agua se atraen entre sí. Debido a esta interacción se forman cuerpos de agua por adhesión de moléculas de agua, las gotas.
Los puentes de hidrógeno mantienen las moléculas de agua fuertemente unidas, formando una estructura compacta que la convierte en un líquido casi incompresible. Al no poder comprimirse puede funcionar en algunos animales como un esqueleto hidrostático, como ocurre en algunos gusanos perforadores capaces de agujerear la roca mediante la presión generada por sus líquidos internos. Estos puentes se pueden romper fácilmente con la llegada de otra molécula con un polo negativo o positivo dependiendo de la molécula, o, con el calor.
A estas rupturas o disociaciones he llamado capa límite hidro química y capa límite térmica.
La fuerza de cohesión permite que el agua se mantenga líquida a temperaturas no extremas.
Adhesión
El agua, por su gran potencial de polaridad, cuenta con la propiedad de la adhesión, es decir, el agua generalmente es atraída y se mantiene adherida a otras superficies.
Tensión superficial
Por su misma propiedad de cohesión, el agua tiene una gran atracción entre las moléculas de su superficie, creando tensión superficial. La superficie del líquido se comporta como una película capaz de alargarse y al mismo tiempo ofrecer cierta resistencia al intentar romperla; esta propiedad contribuye a que algunos objetos muy ligeros floten en la superficie del agua aún siendo más densos que esta. Si colocamos con cuidado una aguja de metal en la superficie de un vaso de agua, veremos cómo flota.
Debido a su elevada tensión superficial, algunos insectos pueden estar sobre ella sin sumergirse e, incluso, hay animales que corren sobre ella, como el basilisco. También es la causa de que se vea muy afectada por fenómenos de capilaridad.
Las gotas de agua son estables también debido a su alta tensión superficial. Esto se puede ver cuando pequeñas cantidades de agua se ponen en superficies no solubles, como el vidrio, donde el agua se agrupa en forma de gotas.
Acción capilar
El agua cuenta con la propiedad de la capilaridad, que es la propiedad de ascenso, o descenso, de un líquido dentro de un tubo capilar. Esto se debe a sus propiedades de adhesión y cohesión.
Cuando se introduce un capilar en un recipiente con agua, ésta asciende espontáneamente por el capilar como si trepase "agarrándose" por las paredes, hasta alcanzar un nivel superior al del recipiente, donde la presión que ejerce la columna de agua se equilibra con la presión capilar. A este fenómeno se debe, en parte, la ascensión de la savia bruta, desde las raíces hasta las hojas, a través de los vasos leñosos.
Calor específico
Esta propiedad también se encuentra en relación directa con la capacidad del agua para formar puentes de hidrógeno intermoleculares. El agua puede absorber grandes cantidades de calor que es utilizado para romper los puentes de hidrógeno, por lo que la temperatura se eleva muy lentamente.
Bastan mínimas diferencias térmicas para romper ese enlace. A esta ruptura o disociación he llamado capa límite térmica. En el eje de esa disociación se manifiesta la descarga de los prolijos bordes cuspidados que van conformando los delicados cordones litorales que asisten las salidas tributarias. La sedimentología, siguiendo el catecismo mecanicistas, aún sigue pensando en olas oblicuas.
El calor específico del agua se define como la cantidad de energía necesaria para elevar la temperatura, en un grado Celsius, a un gramo de agua en condiciones estándar y es de 1 cal/°C•g, que es igual a 4,1840 J/K•g.
Esta propiedad es fundamental para los seres vivos (y la Biosfera en general) ya que gracias a esto, el agua reduce los cambios bruscos de temperatura, siendo un regulador térmico muy bueno. Un ejemplo de esto son las temperaturas tan suaves que hay en las zonas costeras, que son consecuencias de estas propiedad. También ayuda a regular la temperatura de los animales y las células permitiendo que el citoplasma acuoso sirva de protección ante los cambios de temperatura. Así se mantiene la temperatura constante.
La capacidad calorífica del agua es mayor que la de otros líquidos.
Para evaporar el agua se necesita mucha energía. Primero hay que romper los puentes y posteriormente dotar a las moléculas de agua de la suficiente energía cinética para pasar de la fase líquida a la gaseosa. Para evaporar un gramo de agua se precisan 540 calorías, a una temperatura de 20 °C.
Temperatura de fusión y evaporación
Presenta un punto de ebullición de 100 °C (373,15 K) a presión de 1 atmósfera (se considera como estándar para la presión de una atmósfera la presión promedio existente al nivel del mar). El calor latente de evaporación del agua a 100 °C es 540 cal/g (ó 2260 J/g).
Tiene un punto de fusión de 0 °C (273,15 K) a presión de 1 atm. El calor latente de fusión del hielo a 0 °C es 80 cal/g (ó 335 J/g). Tiene un estado de sobre enfriado líquido a −25 °C.
La temperatura crítica del agua, es decir, aquella a partir de la cual no puede estar en estado líquido independientemente de la presión a la que esté sometida, es de 374 °C y se corresponde con una presión de 217,5 atmósferas.
Densidad
La densidad del agua líquida es muy estable y varía poco con los cambios de temperatura y presión. Revisar o enriquecer el concepto de lo picnal en mecánica de fluidos. Revisar asimismo el concepto de viscosidad pues la adherencia electroestática intermolecular afecta su dinámica.
A la presión normal (1 atmósfera), el agua líquida tiene una mínima densidad a los 100 °C, donde tiene 0,958 kg/L. Mientras baja la temperatura, aumenta la densidad (por ejemplo, a 90 °C tiene 0,965 kg/L) y ese aumento es constante hasta llegar a los 4,0 °C donde alcanza una densidad de 1 kg/L. A esa temperatura (4,0 °C) alcanza su máxima densidad (a la presión mencionada). A partir de ese punto, al bajar la temperatura, la densidad comienza a disminuir, aunque muy lentamente, hasta que a los 0 °C disminuye hasta 0,9999 kg/L. Cuando pasa al estado sólido (a 0 °C), ocurre una brusca disminución de la densidad pasando de 0,9999 kg/L a 0,917 kg/L.
Destilación
Para obtener agua químicamente pura es necesario realizar diversos procesos físicos de purificación ya que el agua es capaz de disolver una gran cantidad de sustancias químicas, incluyendo gases.
Se llama agua destilada al agua que ha sido evaporada y posteriormente condensada. Al realizar este proceso se eliminan casi la totalidad de sustancias disueltas y microorganismos que suele contener el agua y el resultado es prácticamente la sustancia química pura H2O.
El agua pura no conduce la electricidad, pues está libre de sales y minerales.
Importancia de la posición astronómica de la Tierra
Si la posición de la Tierra en el Sistema Solar fuera más cercana o más alejada del Sol, la existencia de las condiciones que permiten a las formas del agua estar presentes simultáneamente serían menos probables.
La masa de la Tierra permite mantener la atmósfera. El vapor de agua y el dióxido de carbono en la atmósfera causan el efecto invernadero, lo que ayuda a mantener relativamente constante la temperatura superficial. Si el planeta tuviera menos masa, una atmósfera más delgada causaría temperaturas extremas no permitiendo la acumulación de agua excepto en los casquetes polares
El cambio del estado en el agua
Estado sólido
Al estar el agua en estado sólido, todas las moléculas se encuentran unidas mediante un enlace de hidrógeno, que es un enlace intermolecular y forma una estructura parecida a un panal de abejas, lo que explica que el agua sea menos densa en estado sólido que en el estado líquido. La energía cinética de las moléculas es muy baja, es decir que las moléculas están casi inmóviles.
El agua glacial sometida a extremas temperaturas y presiones criogénicas, adquiere una alta capacidad de sublimación, al pasar de sólida a vapor por la acción energética de los elementos que la integran —oxígeno e hidrógeno— y del calor atrapado durante su proceso de congelación-expansión. Es decir, por su situación de confinamiento a grandes profundidades se deshiela parcialmente, lo cual genera vapor a una temperatura ligeramente superior del helado entorno, suficiente para socavar y formar cavernas en el interior de los densos glaciales. Estas grutas, que además contienen agua proveniente de sistemas subglaciales, involucran a las tres fases actuales del agua, donde al interactuar en un congelado ambiente subterráneo y sin la acción del viento se transforman en el cuarto estado del agua: plasma semilíquido o gelatinoso.
Estado gaseoso
Cuando el agua es gaseosa, la energía cinética es tal que se rompen todos los enlaces de hidrógeno quedando todas las moléculas libres. El vapor de agua es tan invisible como el aire; el vapor que se observa sobre el agua en ebullición o en el aliento emitido en aire muy frío, está formado por gotas microscópicas de agua líquida en suspensión; lo mismo que las nubes

. . . . . . . . . . 
Intermolecular forces (forces between two molecules) are weak compared to the intramolecular forces (forces keeping a molecule together). For example, the covalent bond present within HCl molecules is much stronger than the forces present between the neighbouring molecules. These forces exist between molecules when they are sufficiently close to each other. The forces consist of four types:
- Dipole–dipole forces
- Ion–dipole forces
- Dipole-induced dipole force or Debye forces
- Instantaneous dipole-induced dipole forces or London dispersion forces
London dispersion forces
Otherwise known as quantum-induced instantaneous polarization or instantaneous dipole-induced dipole forces, the London dispersion force is caused by correlated movements of the electrons in interacting molecules. The electrons, which belong to different molecules, start "feeling" and avoiding each other at the short intermolecular distances, which is frequently described as formation of "instantaneous dipoles" that attract each other.
Debye (induced dipole) force
The induced dipole forces appear from the induction (also known as polarization), which is the attractive interaction between a permanent multipole on one molecule with an induced (by the former di/multi-pole) multipole on another (Ref 4-7). This interaction is called Debye force after Peter J.W. Debye.
The example of an induction-interaction between permanent dipole and induced dipole is HCl and Ar. In this system, Ar experiences a dipole as its electrons are attracted (to H side) or repelled (from Cl side) by HCl (Ref 4, 6). This kind of interaction can be expected between any polar molecule and non-polar/symmetrical molecule. The induction-interaction force is far weaker than dipole-dipole interaction, however stronger than London force.They induce their properties in another atom
Dipole–dipole interactions
Dipole–dipole interactions are electrostatic interactions of permanent dipoles in molecules. These interactions tend to align the molecules to increase the attraction (reducing potential energy). An example of a dipole–dipole interaction can be seen in hydrogen chloride (HCl): The positive end of a polar molecule will attract the negative end of the other molecule and cause them to be arranged in a specific arrangement. Polar molecules have a net attraction between them. For example HCl and chloroform (CHCl3)
![]()
Keesom interactions (named after Willem Hendrik Keesom) are attractive interactions of dipoles that are Boltzmann-averaged over different rotational orientations of the dipoles. The energy of a Keesom interaction depends on the inverse sixth power of the distance, unlike the interaction energy of two spatially fixed dipoles, which depends on the inverse third power of the distance.
Often, molecules have dipolar groups within them, but have no overall dipole moment. This occurs if there is symmetry within the molecule, causing the dipoles to cancel each other out. This occurs in molecules such as tetrachloromethane. Note that the dipole–dipole interaction between two atoms is usually zero, because atoms rarely carry a permanent dipole. See atomic dipoles.
Hydrogen bonding
A hydrogen bond is the attractive force between a hydrogen atom and an electronegative atom, that is bonded to either nitrogen, oxygen, or fluorine.[1] The hydrogen bond is often described as a strong electrostatic dipole–dipole interaction. However, it also has some features of covalent bonding: It is directional, stronger than a van der Waals interaction, produces interatomic distances shorter than sum of van der Waals radii, and usually involves a limited number of interaction partners, which can be interpreted as a kind of valence.
Intermolecular hydrogen bonding is responsible for the high boiling point of water (100 °C) compared to the other group 16 hydrides that have no hydrogen bonds. Intramolecular hydrogen bonding is partly responsible for the secondary, tertiary, and quaternary structures of proteins and nucleic acids. It also plays an important role in the structure of polymers, both synthetic and natural.
Relative strength of forces
Bond type |
|
Covalent |
400 |
Hydrogen bonds |
12–16 |
Dipole–dipole |
0.5–2 |
London (van der Waals) Forces |
<1 |
Note: this comparison is only approximate – the actual relative strengths will vary depending on the molecules involved.
In chemistry, a polar bond is a type of covalent bond between two atoms or more in which electrons are shared unequally. Because of this, one end of the molecule has a slight, relative negative charge and the other a slight, relative positive charge. An example of atoms with a polar bond is the water molecule, which is made up of 2 hydrogen atoms and 1 oxygen atom.
In chemistry, polarity refers to a separation of electric charge leading to a molecule or its chemical groups having an electric dipole or multipole moment. Polar molecules interact through dipole–dipole intermolecular forces and hydrogen bonds. Molecular polarity is dependent on the difference in electronegativity between atoms in a compound and the asymmetry of the compound's structure. For example, a molecule of water is polar because of the unequal sharing of its electrons between oxygen and hydrogen in which the former has larger electronegativity than the latter, resulting in a "bent" structure, whereas methane is considered nonpolar because the carbon shares the electrons with the hydrogen atoms almost uniformly. Polarity underlies a number of physical properties including surface tension, solubility, and melting- and boiling-points.
Water-elpot-transparent-3D-balls.pn

The bond dipole moment uses the idea of electric dipole moment to measure the polarity of a chemical bond within a molecule. The bond dipole μ is given by:
![]()
The bond dipole is modeled as +δ — δ- with a distance d between the partial charges +δ and δ-. It is a vector, parallel to the bond axis, pointing from minus to plus, as is conventional[1] for electric dipole moment vectors. (Some chemists draw the vector the other way around, pointing from plus to minus, but only in situations where the direction doesn't matter.)[1] This vector can be physically interpreted as the movement undergone by electrons when the two atoms are placed a distance d apart and allowed to interact, the electrons will move from their free state positions to be localised more around the more electronegative atom.
The SI unit for electric dipole moment is the coulomb-meter, but that is much too large to be practical on the molecular scale. Bond dipole moments are commonly measured in debyes, represented by the symbol D, which is what you get if you measure the charge δ in units of 10-10statcoulomb and measure the distance d in Angstroms. Note that 10-10 statcoulomb is 0.48 units of elementary charge. Another useful conversion factor is 1 C m = 2.9979×1029 D.
Typical dipole moments for simple diatomic molecules are in the range of 0 to 11D. At one extreme, a symmetrical molecule such as chlorine, Cl2, has zero dipole moment, while near the other extreme, gas phase potassium bromide, KBr, which is highly ionic, has a dipole moment of 10.5D.[2]
For a complete molecule the total molecular dipole moment may be approximated as the vector sum of individual bond dipole moments. Often bond dipoles are obtained by the reverse process: a known total dipole of a molecule can be decomposed into bond dipoles. The reason for doing this is the transfer of bond dipole moments to molecules that have the same bonds, but for which the total dipole moment is not yet known. The vector sum of the transferred bond dipoles gives an estimate for the total (unknown) dipole of the molecule.
The Bond Dipole is two atoms in a bond, such that the electronegativity of one atom causes electrons to be drawn towards the other, in turn causing a partial negative charge. There is therefore a difference in polarity across the bond, which causes a dipole moment.
A partial charge is a charge with an absolute value of less than one elementary charge unit (that is, smaller than the charge of the electron).
Partial atomic charges
Partial charges are created due to the asymmetric distribution of electrons in chemical bonds. The resulting partial charges are a property only of zones within the distribution, and not the assemblage as a whole. For example, chemists often choose to look at a small space surrounding the nucleus of an atom: When an electrically neutral atom bonds chemically to another neutral atom that is more electronegative, its electrons are partially drawn away. This leaves the region about that atom's nucleus with a partial positive charge, and it creates a partial negative charge on the atom to which it is bonded.
In such a situation, the distributed charges taken as a group always carries a whole number of elementary charge units. Yet one can point to zones within the assemblage where less than a full charge resides, such as the area around an atom's nucleus. This is possible in part because particles are not like mathematical points--which must be either inside a zone or outside it--but are smeared out by the uncertainty principle of quantum mechanics. Because of this smearing effect, if one defines a sufficiently small zone, a fundamental particle may be both partly inside and partly outside it.
Uses
Partial atomic charges are used in molecular mechanicsforce fields to compute the electrostatic interaction energy using Coulomb's law. They are also often used for a qualitative understanding of the structure and reactivity of molecules.
Methods of determining partial atomic charges
Despite its usefulness, the concept of a partial atomic charge is somewhat arbitrary, because it depends on the method used to delimit between one atom and the next (in reality, atoms have no clear boundaries). As a consequence, there are many methods for estimating the partial charges. According to Cramer (2002), all methods can be classified in one of four classes:
- Class I charges are those that are not determined from quantum mechanics, but from some intuitive or arbitrary approach. These approaches can be based on experimental data such as dipoles and electronegativities.
- Class II charges are derived from partitioning the molecular wave function using some arbitrary, orbital based scheme.
- Class III charges are based on a partitioning of a physical observable derived from the wave function, such as electron density.
- Class IV charges are derived from a semiempirical mapping of a precursor charge of type II or III to reproduce experimentally determined observables such as dipole moments.
In physics, there are several kinds of dipoles:
- An electric dipole is a separation of positive and negative charges. The simplest example of this is a pair of electric charges of equal magnitude but opposite sign, separated by some (usually small) distance. A permanent electric dipole is called an electret.
- A magnetic dipole is a closed circulation of electric current. A simple example of this is a single loop of wire with some constant current flowing through it.[1][2]
- A flow dipole is a separation of a sink and a source. In a highly viscous medium, a two-beater kitchen mixer causes a dipole flow field.
- An acoustic dipole is the oscillating version of it. A simple example is a dipole speaker.
- Any scalar or other field may have a dipole moment.
Dipoles can be characterized by their dipole moment, a vector quantity. For the simple electric dipole given above, the electric dipole moment points from the negative charge towards the positive charge, and has a magnitude equal to the strength of each charge times the separation between the charges. (To be precise: for the definition of the dipole moment one should always consider the "dipole limit", where e.g. the distance of the generating charges should converge to 0, while simultaneously the charge strength should diverge to infinity in such a way that the product remains a positive constant.)
For the current loop, the magnetic dipole moment points through the loop (according to the right hand grip rule), with a magnitude equal to the current in the loop times the area of the loop.
In addition to current loops, the electron, among other fundamental particles, has a magnetic dipole moment. This is because it generates a magnetic field that is identical to that generated by a very small current loop. However, to the best of our knowledge, the electron's magnetic moment is not due to a current loop, but is instead an intrinsic property of the electron.[3] It is also possible that the electron has an electric dipole moment, although this has not yet been observed (see electron electric dipole moment for more information).
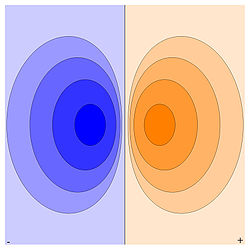
Contour plot of the electrostatic potential of a horizontally oriented electrical dipole of finite size. Strong colors indicate highest and lowest potential (where the opposing charges of the dipole are located).
Molecular dipoles
Many molecules have such dipole moments due to non-uniform distributions of positive and negative charges on the various atoms. Such is the case with polar compounds like hydrogen fluoride (HF), where electron density is shared unequally between atoms.
A molecule with a permanent dipole moment is called a polar molecule. A molecule is polarized when it carries an induced dipole. The physical chemist Peter J. W. Debye was the first scientist to study molecular dipoles extensively, and, as a consequence, dipole moments are measured in units named debye in his honor.
With respect to molecules, there are three types of dipoles:
- Permanent dipoles: These occur when two atoms in a molecule have substantially different electronegativity: One atom attracts electrons more than another, becoming more negative, while the other atom becomes more positive. See dipole-dipole attractions.
- Instantaneous dipoles: These occur due to chance when electrons happen to be more concentrated in one place than another in a molecule, creating a temporary dipole. See instantaneous dipole.
- Induced dipoles: These can occur when one molecule with a permanent dipole repels another molecule's electrons, "inducing" a dipole moment in that molecule. See induced-dipole attraction.
More generally, an induced dipole of any polarizable charge distribution ρ (remember that a molecule has a charge distribution) is caused by an electric field external to ρ. This field may, for instance, originate from an ion or polar molecule in the vicinity of ρ or may be macroscopic (e.g., a molecule between the plates of a charged capacitor). The size of the induced dipole is equal to the product of the strength of the external field and the dipole polarizability of ρ.
Electronegativity, symbol χ (the Greek letter chi), is a chemical property that describes the tendency of an atom or a functional group to attract electrons (or electron density) towards itself and thus the tendency to form negative ions.[1] An atom's electronegativity is affected by both its atomic number and the distance that its valence electrons reside from the charged nucleus. The higher the associated electronegativity number, the more an element or compound attracts electrons towards it. First proposed by Linus Pauling in 1932 as a development of valence bond theory,[2] it has been shown to correlate with a number of other chemical properties. Electronegativity cannot be directly measured and must be calculated from other atomic or molecular properties. Several methods of calculation have been proposed and, although there may be small differences in the numerical values of the electronegativity, all methods show the same periodic trends between elements.
The most commonly used method of calculation is that originally proposed by Linus Pauling. This gives a dimensionless quantity, commonly referred to as the Pauling scale, on a relative scale running from around 0.7 to 3.98 (hydrogen = 2.20). When other methods of calculation are used, it is conventional (although not obligatory) to quote the results on a scale that covers the same range of numerical values: this is known as an electronegativity in Pauling units.
Electronegativity, as it is usually calculated, is not strictly a property of an atom, but rather a property of an atom in a molecule.[3] Properties of a free atom include ionization energy and electron affinity. It is to be expected that the electronegativity of an element will vary with its chemical environment,[4] but it is usually considered to be a transferable property, that is to say that similar values will be valid in a variety of situations.
On the most basic level, electronegativity is determined by factors like the nuclear charge (the more protons an atom has, the more "pull" it will have on negative electrons) and the number/location of other electrons present in the atomic shells (the more electrons an atom has, the farther from the nucleus the valence electrons will be, and as a result the less positive charge they will experience -- both because of their increased distance from the nucleus, and because the other electrons in the lower energy core orbitals will act to shield the valence electrons from the positively charged nucleus).

The opposite of electronegativity is electropositivity: a measure of an element's ability to donate electrons.
Electropositivity is a measure of an element's ability to donate electrons, and therefore form positive ions; thus, it is opposed to electronegativity. Mainly, this is an attribute of metals, meaning that for the most part, the greater the metallic character of an element, the greater the electropositivity. Therefore the alkali metals are most electropositive of all. This is because they have a single electron in their outer shell and, as this is relatively far from the nucleus of the atom, it is easily lost; in other words, these metals have low ionization energies.[24]
While electronegativity increases along periods in the periodic table, and decreases down groups, electropositivity decreases along periods and increases down groups.
Versiones MACRO
The dipole model of the Earth's magnetic field is a first order approximation of the rather complex true Earth's magnetic field. Due to effects of the interplanetary magnetic field, and the solar wind, the dipole model is particularly inaccurate at high L-shells (e.g., above L=3), but may be a good approximation for lower L-shells. For more precise work, or for any work at higher L-shells, a more accurate model that incorporates solar effects, such as the Tsyganenko magnetic field model, is recommended.
The Indian Ocean Dipole (IOD) is an irregular oscillation of sea-surface temperatures in which the western Indian Ocean becomes alternately warmer and then colder than the eastern part of the ocean.
The phenomenon
The IOD involves an aperiodic oscillation of sea-surface temperatures, between "positive", "neutral" and "negative" phases. A positive phase sees greater-than-average sea-surface temperatures and greater precipitation in the western Indian Ocean region, with a corresponding cooling of waters in the eastern Indian Ocean—which tends to cause droughts in adjacent land areas of Indonesia and Australia. The negative phase of the IOD brings about the opposite conditions, with warmer water and greater precipitation in the eastern Indian Ocean, and cooler and drier conditions in the west.
The IOD also affects the strength of monsoons over the Indian subcontinent. A significant positive IOD occurred in 1997-8, with another in 2006. The IOD is one aspect of the general cycle of global climate, interacting with similar phenomena like the El Niño-Southern Oscillation (ENSO) in the Pacific Ocean.
The IOD phenomenon was first identified by climate researchers in 1999. Yet evidence from fossil coral reefs demonstrates that the IOD has functioned since at least the middle of the Holocene period, 6500 years ago.
An average of four each positive/negative IOD events occur during each 30 year period with each event lasting around six months. However, there have been 12 positive IODs since 1980 and no negative events since 1992. The occurrence of consecutive positive IOD events are extremely rare with only two such events recorded, 1913–1914 and the three consecutive events from 2006-2008 which preceded the Black Saturday bushfires. Modelling indicates that consecutive positive events occur twice over a 1,000 year period. The positive IOD in 2007 evolved together with La Niña which is a very rare phenomenon that has happened only once in the available historical records (in 1967).[1][2][3][4]
Effect on Australian Droughts
A 2009 study by Ummenhofer et al. at the University of New South Wales (UNSW) Climate Change Research Centre, has demonstrated a significant correlation between the IOD and drought in the southern half of Australia, in particular the south-east. Every major southern drought since 1889 has coincided with positive/neutral IOD fluctuations including the 1895-1902, 1937–1945 and the current 1995-present droughts.[5]
The research shows that when the IOD is in its negative phase, with cool Indian Ocean water west of Australia and warm Timor Sea water to the north, winds are generated that pick up moisture from the ocean and then sweep down towards southern Australia to deliver higher rainfall. In the IOD positive phase, the pattern of ocean temperatures is reversed, weakening the winds and reducing the amount of moisture picked up and transported across Australia. The consequence is that rainfall in the south-east is well below average during periods of a positive IOD.
The study also shows that the IOD has a much more significant effect on the rainfall patterns in south-east Australia than the El Niño-Southern Oscillation (ENSO) in the Pacific Ocean as already shown in some of the previous [6][7] and recent studies.[8]
Indian Ocean causes Big Dry: drought mystery solved
Bob Beale . February 5, 2009
Dr Caroline Ummenhofer
The causes of south-eastern Australia's longest, most severe and damaging droughts have been discovered, with the surprise finding that they originate far away in the Indian Ocean.
A team of Australian scientists has detailed for the first time how a phenomenon known as the Indian Ocean Dipole (IOD) - a variable and irregular cycle of warming and cooling of ocean water - dictates whether moisture-bearing winds are carried across the southern half of Australia.
The landmark new study explains the current record-breaking drought in south-eastern Australia and solves the mystery of why a string of La Nina events in the Pacific Ocean - which usually bring rain - has failed to break it.
It also reveals the causes of other iconic extreme droughts in recorded history, notably the World War II Drought from 1937 to 1945 and the Federation Drought from 1895 to 1902, and challenges the accepted understanding of the key drivers of Australia's climate.
When the IOD is in its negative phase, a pattern occurs with cool Indian Ocean water west of Australia and warm Timor Sea water to the north. This generates winds that pick up moisture from the ocean and then sweep down towards southern Australia to deliver wet conditions. In its positive phase, the pattern of ocean temperatures is reversed, weakening the winds and reducing the amount of moisture picked up and transported across Australia. So the south-east misses out on its usual quota of rain.
The study notes that the IOD has been in its positive or neutral phase since 1992, the longest period of its kind since records began in the late 19th Century.

How the Indian Ocean drives Australia's worst droughts. Click image to enlarge.
To make matters worse, this period has coincided with a trend towards higher average air temperatures over the land, which may be linked to human-induced climate change.
The team, led by Dr Caroline Ummenhofer and Professor Matthew England of the UNSW Climate Change Research Centre, details its findings in a paper accepted for publication in the journal Geophysical Review Letters. The team included researchers from CSIRO Centre for Australian Weather and Climate Research and the University of Tasmania.
"The ramifications of drought for this region are dire, with acute water shortages for rural and metropolitan areas, record agricultural losses, the drying-out of two of Australia's major river systems and far-reaching ecosystem damage," says Dr Ummenhofer.
"We have shown that the state of the Indian Ocean is highly important for rainfall and droughts in south-east Australia. More than the variability associated with the El Nino/La Nina cycle in the Pacific Ocean, the Indian Ocean Dipole is the key factor for driving major south-east Australian droughts over the past 120 years.

Top graph shows phases of Indian Ocean Dipole back to
1880. Normal rain declines in positive phase. Grey bars
indicate drought. Bottom graph shows rainfall
anomalies (mm). Click image to enlarge.
"During this latest drought - the so-called 'Big Dry' - recent higher air temperatures across south-eastern Australia have exacerbated the problem.
"Our findings will help to improve seasonal rainfall forecasts and therefore directly benefit water and agricultural management."

MACRO. Con nula energía gravitacional; cargados de energías convectivas; el Paraná y el Paraguay se aparean en Paso de la Patria y conservan disociación térmica e hidroquímica hasta alcanzar las aguas del Plata.

Al esfuerzo por relacionar energía y materia van estos textos que siguen. Menos mal que los fenómenos entran por los ojos y no por la razón.
Quantum mechanical theories
Quantum-mechanical explanation of intermolecular interactions
Perturbation theory
Hydrogen bonding, dipole–dipole interactions, and London (Van der Waals) forces are most naturally accounted for by Rayleigh–Schrödinger perturbation theory (RS-PT). In this theory—applied to two monomers A and B—one uses as unperturbed Hamiltonian the sum of two monomer Hamiltonians, ![]()
In the present case the unperturbed states are products ![]() with
with ![]() and
and ![]()
Supermolecular approach
The early theoretical work on intermolecular forces was invariably based on RS-PT and its antisymmetrized variants. However, since the beginning of the 1990s it has become possible to apply standard quantum chemical methods to pairs of molecules. This approach is referred to as the supermolecule method. In order to obtain reliable results one must include electronic correlation in the supermolecule method (without it dispersion is not accounted for at all), and take care of the basis set superposition error. This is the effect that the atomic orbital basis of one molecule improves the basis of the other. Since this improvement is distance dependent, it easily gives rise to artifacts.
Exchange
The monomer functions ΦnA and ΦmB are antisymmetric under permutation of electron coordinates (i.e., they satisfy the Pauli principle), but the product states are not antisymmetric under intermolecular exchange of the electrons. An obvious way to proceed would be to introduce the intermolecular antisymmetrizer![]() . But, as already noticed in 1930 by Eisenschitz and London,[1] this causes two major problems. In the first place the antisymmetrized unperturbed states are no longer eigenfunctions of H(0), which follows from the non-commutation
. But, as already noticed in 1930 by Eisenschitz and London,[1] this causes two major problems. In the first place the antisymmetrized unperturbed states are no longer eigenfunctions of H(0), which follows from the non-commutation
![]()
In the second place the projected excited states
![]()
become linearly dependent and the choice of a linearly independent subset is not apparent. In the late 1960s the Eisenschitz–London approach was revived and different rigorous variants of symmetry adapted perturbation theory were developed. (The word symmetry refers here to permutational symmetry of electrons). The different approaches shared a major drawback: they were very difficult to apply in practice. Hence a somewhat less rigorous approach (weak symmetry forcing) was introduced: apply ordinary RS-PT and introduce the intermolecular antisymmetrizer at appropriate places in the RS-PT equations. This approach leads to feasible equations, and, when electronically correlated monomer functions are used, weak symmetry forcing is known to give reliable results.[2][3]
The first-order (most important) energy including exchange is in almost all symmetry-adapted perturbation theories given by the following expression

The main difference between covalent and non-covalent forces is the sign of this expression. In the case of chemical bonding this interaction is attractive (for certain electron-spin state, usually spin-singlet) and responsible for large bonding energies—on the order of a hundred kcal/mol. In the case of intermolecular forces between closed shell systems, the interaction is strongly repulsive and responsible for the "volume" of the molecule (see Van der Waals radius). Roughly speaking, the exchange interaction is proportional to the differential overlap between Φ0A and Φ0B. Since the wavefunctions decay exponentially as a function of distance, the exchange interaction does too. Hence the range of action is relatively short, which is why exchange interactions are referred to as short range interactions.
Electrostatic interactions
By definition the electrostatic interaction is given by the first-order Rayleigh–Schrödinger perturbation (RS-PT) energy (without exchange):
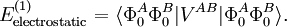
Let the clamped nucleus α on A have position vector Rα, then its charge times the Dirac delta function, Zαδ(r − Rα), is the charge density of this nucleus. The total charge density of monomer A is given by
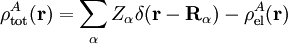
with the electronic charge density given by an integral over nA − 1 primed electron coordinates:

An analogous definition holds for the charge density of monomer B. It can be shown that the first-order quantum mechanical expression can be written as

which is nothing but the classical expression for the electrostatic interaction between two charge distributions. This shows that the first-order RS-PT energy is indeed equal to the electrostatic interaction between A and B.
Multipole expansion
At present it is feasible to compute the electrostatic energy without any further approximations other than those applied in the computation of the monomer wavefunctions. In the past this was different and a further approximation was commonly introduced: VAB was expanded in a (truncated) series in inverse powers of the intermolecular distance R. This yields the multipole EXPANSION of the electrostatic energy. Since its concepts still pervade the theory of intermolecular forces, it will be presented here. In this article the following expansion is proved

with the Clebsch–Gordan series defined by

and the irregular solid harmonic is defined by

The function YL,M is a normalized spherical harmonic, while
![]() and
and ![]() are spherical multipole moment operators. This expansion is manifestly in powers of 1/RAB.
are spherical multipole moment operators. This expansion is manifestly in powers of 1/RAB.
Insertion of this expansion into the first-order (without exchange) expression gives a very similar expansion for the electrostatic energy, because the matrix element factorizes,

with the permanent multipole moments defined by
![]()
We see that the series is of infinite length, and, indeed, most molecules have an infinite number of non-vanishing multipoles. In the past, when computer calculations for the permanent moments were not yet feasible, it was common to truncate this series after the first non-vanishing term.
Which term is non-vanishing, depends very much on the symmetry of the molecules constituting the dimer. For instance, molecules with an inversion center such as a homonuclear diatomic (e.g., molecular nitrogen N2), or an organic molecule like ethene (C2H4) do not possess a permanent dipole moment (l=1), but do carry a quadrupole moment (l=2). Molecules such a hydrogen chloride (HCl) and water (H2O) lack an inversion center and hence do have a permanent dipole. So, the first non-vanishing electrostatic term in, e.g., the N2—H2O dimer, is the lA=2, lB=1 term. From the formula above follows that this term contains the irregular solid harmonic of order L = lA + lB = 3, which has an R−4 dependence. But in this dimer the quadrupole-quadrupole interaction (R−5) is not unimportant either, because the water molecule carries a non-vanishing quadrupole as well.
When computer calculations of permanent multipole moments of any order became possible, the matter of the convergence of the multipole series became urgent. It can be shown that, if the charge distributions of the two monomers overlap, the multipole expansion is formally divergent
Ionic interactions
It is debatable whether ionic interactions are to be seen as intermolecular forces; some workers consider them rather as special kind of chemical bonding. The forces occur between charged atoms or molecules (ions).
Ionic bonds are formed when the difference between the electron affinity of one monomer and the ionization potential of the other is so large that electron transfer from the one monomer to the other is energetically favorable. Since a transfer of an electron is never complete there is always a degree of covalent bonding.
Once the ions (of opposite sign) are formed, the interaction between them can be seen as a special case of multipolar attraction, with a 1/RAB distance dependence. Indeed, the ionic interaction is the electrostatic term with lA = 0 and lB = 0. Using that the irregular harmonics for L = 0 is simply

and that the monopole moments and their Clebsch–Gordan coupling are
![]()
(where qA and qB are the charges of the molecular ions) we recover—as to be expected—Coulomb's law

For shorter distances, where the charge distributions of the monomers overlap, the ions will repel each other because of inter-monomer exchange of the electrons.
Ionic compounds have high melting and boiling points due to the large amount of energy required to break the forces between the charged ions. When molten they are also good conductors of heat and electricity, due to the free or delocalized ions.
Writing

and similarly for B, we get the well-known expression

As a numerical example we consider the HCl dimer depicted above. We assume that the left molecule is A and the right B, so that the z-axis is along the molecules and points to the right. Our (physical) convention of the dipole moment is such that it points from negative to positive charge. Note parenthetically that in organic chemistry the opposite convention is used. Since organic chemists hardly ever perform vector computations with dipoles, confusion hardly ever arises. In organic chemistry dipoles are mainly used as a measure of charge separation in a molecule. So,

The value of μHCl is 0.43 (atomic units), so that at a distance of 10 bohr the dipole-dipole attraction is −3.698 10−4 hartree (−0.97 kJ/mol).
If one of the molecules is neutral and freely rotating, the total electrostatic interaction energy becomes zero. (For the dipole-dipole interaction this is most easily proved by integrating over the spherical polar angles of the dipole vector, while using the volume element sinθ dθdφ). In gases and liquids molecules are not rotating completely freely—the rotation is weighted by the Boltzmann factor exp(−Edip-dip/kT), where k is the Boltzmann constant and T the absolute temperature. It was first shown by John Lennard-Jones[4] that the temperature-averaged dipole-dipole interaction is

Since the averaged energy has an R−6 dependence, it is evidently much weaker than the unaveraged one, but it is not completely zero. It is attractive, because the Boltzmann weighting favors somewhat the attractive regions of space. In HCl-HCl we find for T = 300 K and RAB = 10 bohr the averaged attraction −62 J/mol, which shows a weakening of the interaction by a factor of about 16 due to thermal rotational motion.
Anisotropy and non-additivity of intermolecular forces
Consider the interaction between two electric point charges at position ![]() and
and ![]() . By Coulomb's law the interaction potential depends only on the distance
. By Coulomb's law the interaction potential depends only on the distance ![]() between the particles. For molecules this is different. If we see a molecule as a rigid 3-D body, it has 6 degrees of freedom (3 degrees for its orientation and 3 degrees for its position in R3). The interaction energy of two molecules (a dimer) in isotropic and homogeneous space is in general a function of 2×6−6=6 degrees of freedom (by the homogeneity of space the interaction does not depend on the position of the center of mass of the dimer, and by the isotropy of space the interaction does not depend on the orientation of the dimer). The analytic description of the interaction of two arbitrarily shaped rigid molecules requires therefore 6 parameters. (One often uses two Euler angles per molecule, plus a dihedral angle, plus the distance.)
between the particles. For molecules this is different. If we see a molecule as a rigid 3-D body, it has 6 degrees of freedom (3 degrees for its orientation and 3 degrees for its position in R3). The interaction energy of two molecules (a dimer) in isotropic and homogeneous space is in general a function of 2×6−6=6 degrees of freedom (by the homogeneity of space the interaction does not depend on the position of the center of mass of the dimer, and by the isotropy of space the interaction does not depend on the orientation of the dimer). The analytic description of the interaction of two arbitrarily shaped rigid molecules requires therefore 6 parameters. (One often uses two Euler angles per molecule, plus a dihedral angle, plus the distance.)
The fact that the intermolecular interaction depends on the orientation of the molecules is expressed by stating that the potential is anisotropic. Since point charges are by definition spherical symmetric, their interaction is isotropic. Especially in the older literature, intermolecular interactions are regularly assumed to be isotropic (e.g., the potential is described in Lennard-Jones form, which depends only on distance).
Consider three arbitrary point charges at distances r12, r13, and r23 apart. The total interaction U is additive; i.e., it is the sum
U = u(r12) + u(r13) + u(r23).
Again for molecules this can be different. Pretending that the interaction depends on distances only—but see above—the interaction of three molecules takes in general the form
U = u(r12) + u(r13) + u(r23) + u(r12,r13,r23),
where u(r12,r13,r23) is a non-additive three-body interaction. Such an interaction can be caused by exchange interactions, by induction, and by dispersion (the Axilrod–Teller triple dipole effect).
. . . . . . . . . . . . . . . . . . 
Homenaje a los flujos convectivos internos naturales positivos por Blas Castagna
Distinguido Francisco:
Gracias nuevamente por compartir conmigo sus reflexiones e inquietudes.
En el presente caso le escribo porque me gustaría dialogar con usted el significado del concepto de advercción. Dicho término, posee según ciertos sesgos disciplinares definiciones diferentes. Asimismo observo en las conceptualizaciones que ofrece la literatura al respecto, la existencia limites poco claros con otros vocablos como son convección, dispersión, difusión; complicándose más aún cuando se incorpora en los textos el concepto de calor.
En virtud de lo expresado, entiendo que definiciones precisas de su parte en dichos términos, nos esclarecerán y así podremos seguir discutiendo con un lenguaje mas ajustado en pos de profundizar nuestra reflexiones conjuntas.
Haciendo explicita mi admiración y reconocimiento por sus comprometidos quehaceres, lo saludo con entusiasmo, como así también, le manifiesto mis respeto a sus musas.
Román, 3 de Julio del 2017
Román Segovia, Geólogo, doctor en Ciencias Naturales. Observador de las concepciones de procesos geológicos que se ponen en juego en la enseñanza
Sin duda mi muy Estimado Román estas voces que responden a procesos termodinámicos son instaladas en contextos mecánicos y por la mayor difusión de sus esquemas simples hace que su divulgación divague en imprecisiones..
Por eso lo primero que rescato es su pertenencia a sistemas complejos; en este caso descubriendo a nivel molecular intercambios verticales en organizaciones de prismas hexagonales con intercambio de energía que desciende por el centro y asciende por los bordes de estos prismas. A este intercambio vertical se lo denomina convección.
Pero al desplazamiento en horizontal de estos mismos sistemas convectivos les cabe otra denominación: advección. En principio de advertencias, esa advección está determinada por un gradiente de ligera menor temperatura, no mayor a 0,2º. Por cierto, ésto no se agota en ese gradiente.
Las explicaciones a nivel molecular que he leído sobre estos procesos apuntan a una amplia y en mi conciencia no bien precisada variedad de enlaces: covalentes, puentes de hidrógeno, dipolo-dipolo, fuerzas London (van der Waals), como los más “conocidos”.
Los enlaces covalentes son por lejos los más importantes. Luego le siguen en importancia los puentes de hidrógeno.
¿Cómo se relacionan esas organizaciones de prismas hexagonales verticales (sistema convectivo), con estas energías de enlaces intermoleculares determinantes del desplazamiento horizontal, es tema de una complejidad que escapa a mi burro.
Cuando la investigación se disponga a desarrollar nano dispositivos para estudiar estos procesos en el ambiente natural, se irán afinando estas percepciones. Por un lado tienen la ventaja de que son procesos que se descubren en grandes escalas y con ello los problemas de borde se minimizan o desaparecen. En laboratorio es mucho más complejo modelizarlos.
Pero esta etapa ya corresponde a la ciencia.
La primera es fruto de la observación por imagen satelital o aérea de sistemas naturales. Ella es la que abre los sentidos y va paso a paso permitiendo formular fenomenologías y prudentes conceptualizaciones sobre estos enlaces.
La tarea de mis Musas ha sido abrirme la atención a estos fenómenos; que aunque complejos me permiten estimar morfologías y diferenciar lo básico de un proceso eólico, de uno fluvial. Algo para empezar a palpar abismos de criterios errados.
El ojo mecánico es tan simple como presto en afirmaciones. Y así es como la geología, siguiendo ese camino ha errado aprecios que nos dejan alelados.
Por este http://www.alestuariodelplata.com.ar/agua3.html tendrá un breve y presto ingreso a esta complejidad que escapa a ambos, aunque sin embargo, nos alerta de que hemos estado mirando por modelos en extremo simples. Y así es como inferimos energías gravitacionales en planicies con pendientes de 4 mm/km. De ahí en más cualquier error ya tiene invitación a tomar ese camino, que con una pincelada de abstracción cierra el largo destino que tiene la mecánica de fluidos recorrido..
Estos textos son solo alertas, que facilitan tomar el sendero de la observación de los flujos ribereños, de las interfaces tributarias, de las pendientes en los fondos, de los esteros aledaños, de las costas blandas y bordes lábiles de transferencias termodinámicas, de los meandros, de los perfiles transversales bien diferenciados que descubren esos meandros y así en más vamos desarrollandosensibilidad para mirar estos temas sin necesidad de salir de nuestro cómodo lugar de observación.
La tentación de ir en directo a la física matemática para modelar estos procesos, es tan común como indicadora de ausencias de esa sensibilidad previa que decide la tarea de seguir mirando y no esperar milagros con sello matemático.
Por cierto que no espero que sus decisiones surjan de estas explicaciones. Pero si espero que crezcan sus dudas de tantos modelos acreditados en interminable sucesión de fracasos que por todos lados chocan con explicaciones que solo un ciego aceptaría.
Sin las miles de imágenes que asisten estos trabajos, no sabría cómo empezar a explicar los fenómenos que veo. Ls imágenes pareciera que vienen dispuestas a alterar todos nuestros preconceptos. Todos los días nos meten un gol en contra de lo estatuído por las leyes. Tal el caso de la 2ª ley de la termodinámica, tan celebrada como la biblia y tan torpe como esas 0,2º que bastan para destrozarla.
Un abrazo y también a Ud Estimado Román le asistan cada día sospechas sus Musas.
Francisco, 23 de Mayo del 2007 - 3 de Julio del 2017